化学科学与工程学院徐涛课题组基于外源自由基制备磷自由基中间体实现醇的直接脱氧转化,研究成果发表于《德国应用化学》
来源:化学科学与工程学院
时间:2024-02-22 浏览:
醇化合物来源广泛、价格低廉,是一种理想的替代有机卤代物作为C(sp3)源的偶联试剂。然而,由于C-O键极高的解离能,以及羟基的弱离去能力,醇的转化一般依赖于额外的预活化步骤,例如转化为草酸盐或黄原酸酯等。目前已有一些醇脱氧转化的途径,但其往往适应于某一类独特的醇结构,具有普适的醇脱氧转化的方法还十分有限。
利用RO-C碳中心自由基β-断裂产生烷基自由基实现醇的脱氧偶联已有报道,但形成这种中间体需要昂贵的活化试剂,而通过RO-P膦中心自由基β-断裂产生烷基自由基有望提供一种高效活化醇脱氧偶联的方式。近日,同济大学化学科学与工程学院徐涛教授课题组发展了一种基于外源自由基加成得到磷自由基中间体实现醇的直接脱氧转化的新方法,相关研究成果“Deoxygenative Transformation of Alcohols via Phosphoranyl Radical from Exogenous Radical Addition”在线发表于化学领域著名国际学术期刊《德国应用化学》(Angewandte Chemie International Edition)。

研究人员以醇作为反应前体,利用廉价易得的二苯基氯膦作为原位活化试剂,以碘作为外源自由基来源,得到的膦自由基中间体β-断裂产生烷基自由基,一步完成了对醇的原位脱氧活化,高效经济的实现了C(sp3)- C(sp3)和C(sp3)- C(sp2)的构筑。
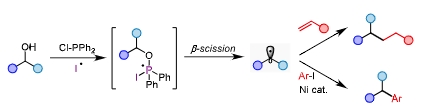
利用这种方法,可以方便的实现一级醇、二级醇、三级醇的烷基化和芳基化。反应体系可以兼容丰富的官能团以及杂环结构,同时能够实现对复杂天然产物和药物的后修饰以及关键中间体的高效合成。利用商业化的氘代甲醇亦可以实现烷基或芳基的氘代甲基化反应,在药物化学上具有一定的应用价值。
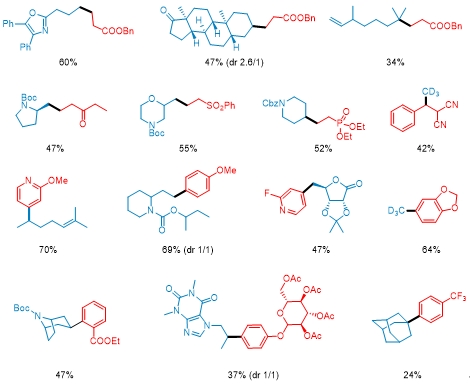
该新型策略可以实现多羟基化合物的选择性转化,同时用于快速获得先前方法难以获得的重要药物中间体中,因此有望在有机合成化学领域发挥重要价值。
徐涛教授课题组一直致力于光镍双催化的不对称还原偶联反应以及羰基去氧双官能团化反应,该研究成果为课题组进一步实现卤代烃偶联试剂的绿色替代、发展醇的脱氧不对称成键反应,以及碳氧键转化反应奠定了重要的基础。
博士研究生徐文豪为论文第一作者,徐涛教授为论文通讯作者。该项研究工作得到了国家自然科学基金、小米青年学者基金等项目的资助。论文得到了洛桑联邦理工大学胡喜乐教授和樊超博士的大力支持。
论文链接:https://doi.org/10.1002/anie.202401575



